

In the previous entry we learned a bit about the “historical” context of the origin of animals, both in the evolutionary and the chronological meaning of the word. We saw how the fossil record was the first resource explored to unraveling the origin of animals, and how it helped to pinpoint the geological period when this lineage emerged. We also learned that comparing the genomes of different animal species has become a new paradigm of research, as it helps to clarify the phylogenetic relationships of animals, as well as it allows to infer the what genetic information needed to build animal bodies was present in the ancestor.
In this entry, we will explore what was the gene content like in the animal LCA in two ways: firstly, by broadly looking at the changes occurred in the genomes of animals during their evolutionary history; secondly, by looking at the genes found scattered across the early branching animals, which together are like pieces of the puzzle that is the animal LCA.
2.1 Genomic trends orchestrating animal evolution
As shown in the review, several independent studies have recently addressed animal genome evolution and the genomic features of the animal LCA by looking at how gene families are distributed across the tree of life using a wide range of genomes. There seems to be a differential enrichment of gene gains and duplications at the stem lineage of animals compared to their unicellular relatives and to other ancestors, accompanied by a largely equal number of losses of gene families that evolved along the animal stem.
Some of the new gene families take part in processes that tell animals apart, such as development, but not all the genes originated in the animal LCA are still present in all extant phyla. Only around 2% of these gene families are conserved across all phyla, a small set that is broadly enriched in functions of DNA binding, Transcription Factors (TFs) and innate immunity (de Mendoza et al., 2013, Paps and Holland, 2018; Richter et al., 2018). Interestingly, members of the Wnt and TGF-β signalling pathways, considered hallmarks of animal development and multicellularity, are well represented in all but highly derived early-branching organisms (Chang et al., 2015; Schenkelaars et al., 2017, Adamska et al., 2007; Lee et al., 2006; Leininger et al., 2014; Paps and Holland, 2018; Richter et al., 2018).
Besides gene innovation, pre-existing gene families expanded and new domain architectures were acquired during the emergence of animals, including receptor tyrosine-kinases, SNARE receptors, and homeobox and helix-loop-helix TFs (de Mendoza et al., 2013; Paps and Holland, 2018; Srivastava et al., 2010; Suga et al., 2013, 2014). These genomic expansions and subfunctionalization are likely related to the increase in complexity in animals, as suggested in different nodes of the metazoan tree of life (de Mendoza et al., 2013; Larroux et al., 2007, 2006; Srivastava et al., 2010, Marletaz et al., 2017, Larroux et al., 2008).
Gene losses also impacted the evolution of animals, with as many gene losses as gene gains in animals compared to their unicellular relatives, affecting pathways such as amino acid biosynthesis and osmosensing (Richter et al., 2018). A remarkable amount of gene loss contributed to shaping genome composition especially during the evolution of two major groups of bilaterian animals and in several deuterostome lineages (Guijarro-Clarke et al., 2020).
Overall, metazoan genomes evolved from a combination of ancient gene families with newly evolved genes in the animal stem lineage, shaped by an unbalanced distribution of gene gain and duplications, rampant gene family losses, gene co-option, and protein domain rearrangement, shuffling and repurposing, that led to subfunctionalization and an increase in regulatory versatility (Fernández and Gabaldón, 2020; Albalat and Cañestro, 2016; Richter et al., 2018; Paps and Holland, 2018; Grau-Bové et al., 2017).
2.2 The animal LCA gene toolkit
What can these studies tell us about the common gene repertoire of all animals? From the aforementioned cases of gains, expansions, and losses, we can conclude that genes related to cell adhesion, motility, and developmental and nervous signaling were already present in the animal LCA. Below follows a detailed overview of what has been found by looking at the genome content from early branching metazoans.
Cell adhesion and organism motility
For example, at the cell adhesion level, cadherins (molecules mediating cell-cell interactions), integrins (mediating cell-extracellular matrix interactions) and some basal lamina elements are found in most non-bilaterians (Moroz et al., 2014; Putnam et al., 2007; Ryan et al., 2013; Srivastava et al., 2010; Fidler et al., 2017), and adherens junction/cell polarity components are fairly well conserved in sponges (Fahey and Degnan, 2010; Nichols et al., 2012; Srivastava et al., 2010) with some homologs missing in ctenophores (Belahbib et al., 2018). These results point to a rich repertoire of adhesion genes in the animal LCA.
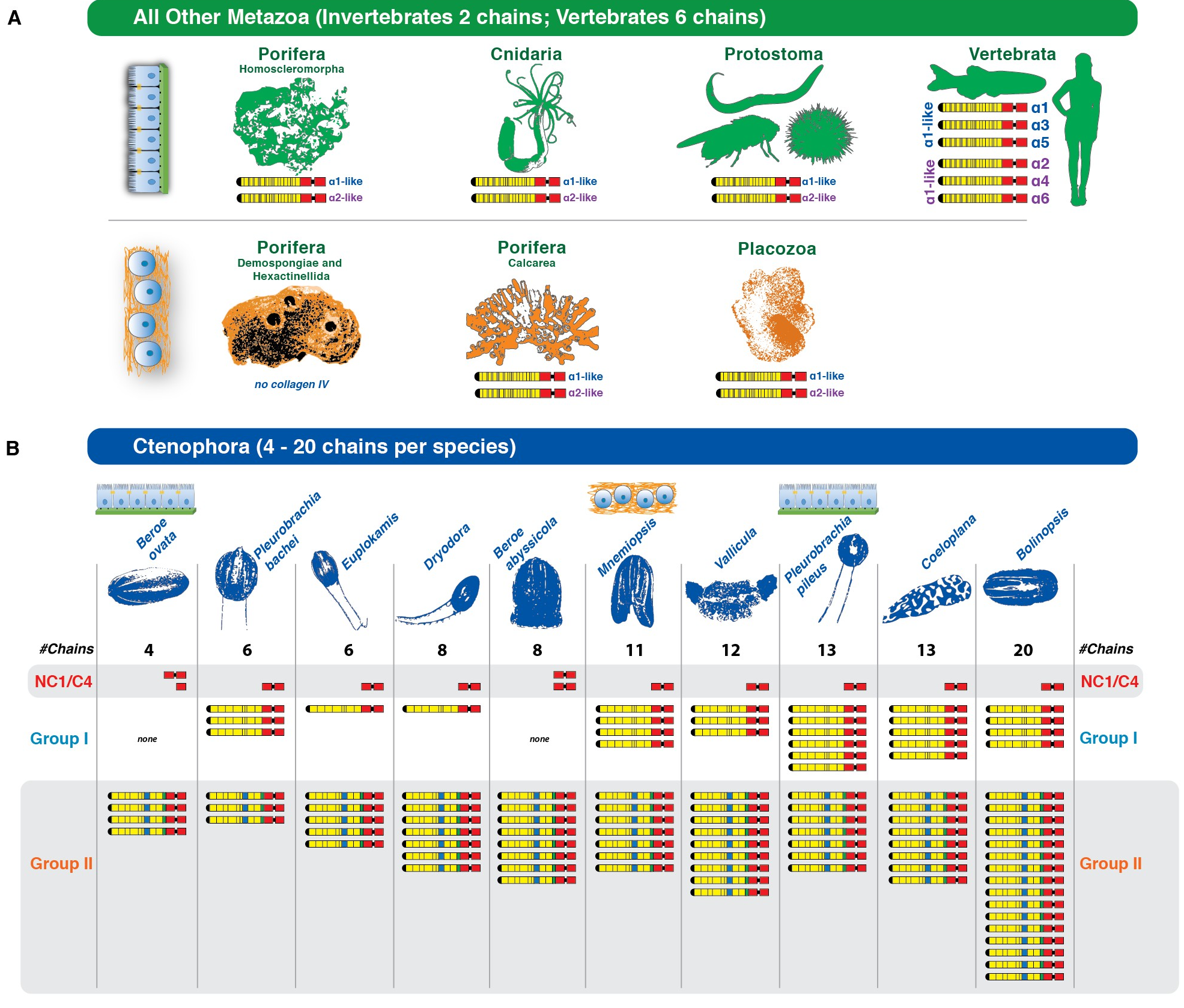
Genes involved in motility have also been found in early-branching animals. For example, although sponges are sessile during adulthood, two orthologs of heavy chain myosin of striated and non-striated muscle have been identified in two sponge genomes, suggesting that a contractile system may have existed in the animal LCA and later evolved into distinct muscle types in different lineages. This system was potentially lost in sponges and independently gave rise to striated muscle cells in cnidarians, ctenophores, and bilaterians (Burton, 2008; Steinmetz et al., 2012).
Nervous system and nerve impulse
Most animals feature the integration of stimuli and the elaboration of responses in the shape of complex behaviors thanks to the nervous system. The LIM+homeobox family of transcription factors was found to be only in metazoa (Putnam 2007 Nvec), and together with genes such as bHLH, sox and elav, lie among the first genes found in animal genomes related to neural development and neural cell fate. Surprisingly, LIM-homeobox genes are present and expressed in developing Amphimedon embryos, despite lacking any overt nervous system (Srivastava et al. 2010a). This same species owns genes related to postsynaptic densities, as well as different receptors and biosynthesis enzymes of different molecules used as transmitters, the latter linked to the production of secondary metabolites (Riesgo et al., 2014). This representation of genes related to the nervous system is however limited in some lineages such as ctenophores, where the main messengers used in synapsis in other animals are absent (Moroz et al., 2014). In sponges, interestingly, it has been documented that many genes associated with neuronal processes are expressed in different cell types, with little evidence as to if these are contributing to some sort of coordination. Some findings hint at the possibility that these genes are performing other roles in sponges, as a result of an early specialization of the neuronal-to-be machinery for filter-feeding organismal strategies (Leys, 2015; Mah and Leys, 2017).
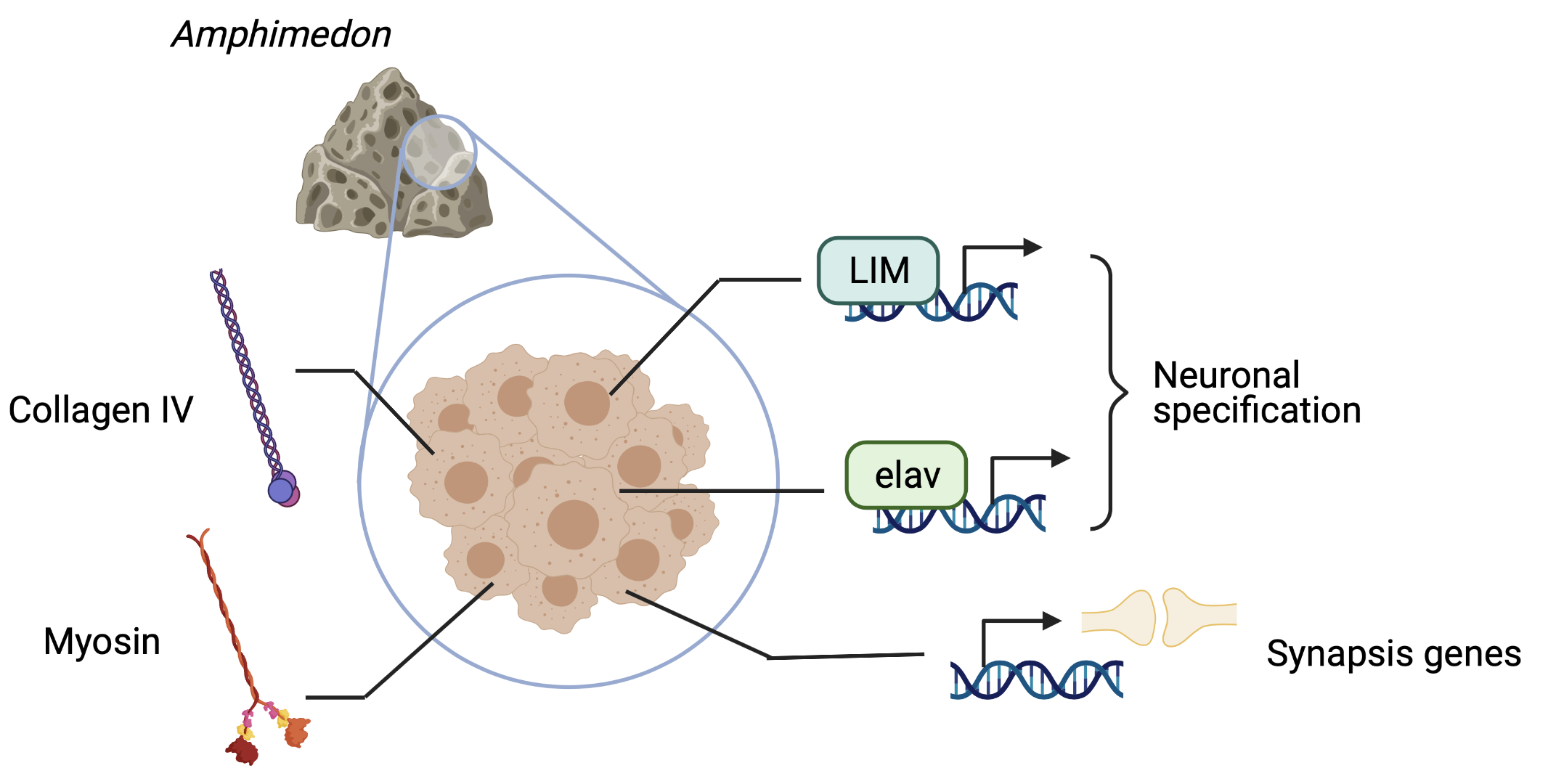
Together with an overall absence of pan-neuronal and pan-synaptic genes across metazoa, (Moroz & Kohn, 2015), this might indicate a parallel evolution of the nervous system in early branching animals such as ctenophores (Moroz & Kohn, 2016). This possibility should not pose the origin of nerve cells as an impossible mystery, however. We can find some of the first hints in the developmental history of nerve cells, whose originating germ layer, the ectoderm, is shared with the perhaps most ancestral of cell types: skin cells. There are numerous examples in nature of electrically excitable epithelia (Josephson, 2004; Mackie, 2004; Roberts, 2004), which suggests a potential common origin of the electrochemical basis observed in nerve impulses and that of epithelia. A machinery for osmotic control (such as usage of ion channels to generate action potential, and a functional basal lamina) in a continuum of cells delimiting compartments is shared between the two tissue types, and was needed prior to the evolution of other neuronal features since they rely on current transmission derived from osmotic changes (Bucher & Anderson, 2015).
Kinship recognition
Recognition of the organism versus the environment is present in all animals. There are numerous evidences of innate immunity components occurring at different animal lineages, from Toll-like and Ig receptors to transcription factors and complement system in sponges and cnidarians (Gauthier et al., 2010, Riesgo et al., 2014, Miller et al., 2007, Brennan et al., 2017). On the contrary, it seems that the mechanisms of allorecognition -the ability to distinguish self and non-self elements within the organism- show no homology between those lineages where they have been described (Grice et al., 2017, Rosengarten and Nicotra, 2011, Karadge et al., 2015, Zárate-Potes et al., 2019). It is needed more research on how allorecognition works in other early-branching organisms in order to infer whether an ancestral machinery of kinship recognition was present in the animal LCA.
Development
All animal structures are organized thanks to a developmental plan regulated with a developmental gene toolkit. Several components of the Wnt signalling pathway, previously discussed as a metazoan innovation, are expressed in sponge larvae, during cnidarian development, and in several structures of both adult sponges and adult ctenophores (Adamska et al., 2007; Hobmayer et al., 2000; Lee et al., 2006; Riesgo et al., 2014; Srivastava et al., 2010; Windsor Reid et al., 2018). Components of other signalling pathways, such as Hedgehog, Notch, or TGF-β, are present but more scattered between lineages and species (Moroz et al., 2014; Riesgo et al., 2014; Ryan et al., 2013). Despite being patchy and incomplete in different species, all animal signalling pathways (including those responsible for patterning bilaterians) are present in virtually all early lineages (Nichols et al., 2006; Paps and Holland, 2018; Riesgo et al., 2014), suggesting the animal LCA contained genetic machinery with the potential to drive axial patterning in a developmental program.
A similar scattering is observed with developmental germ layers. Ctenophores possess an independently-derived mesodermal tissue, despite their lack of key bilaterian mesoderm specification genes (Martindale and Henry, 1999; Moroz et al., 2014; Ryan et al., 2013). This suggests that the regulatory mechanisms necessary for establishing early fates in layers of cells (such as the muscle cells in the ctenophore-specific mesoderm) were present before the emergence of bilaterians. If ctenophores are the earliest branching animals, then these mechanisms would likely have been present in the animal LCA.
Comparison with other distant lineages
The logical immediate step on the quest for genomes places us in deeper roots of the animal tree of life. As thoroughly commented on our review, many of the “unicellular” relatives of animals (a heterogeneous group of lineages with different multicellular strategies) possess genes encoding for numerous “animal-like” features, including developmental signaling genes, cell adhesion genes, diverse genomic regulators, and even genes for synaptic-related processes (Ros-Rocher et al., 2021). Some of these genes, such as integrins, can be found in even deeper roots of the eukaryotic tree of life, down to amoebozoa (see the works of Matt Brown group, for great insights). Thus, the origins of some components essential for animals as we know them are now placed throughout their whole evolutionary history as eukaryotes. Such is the power of genomics.
Comparisons have also been made between animals and other multicellular life forms, although these are complicated by the long evolutionary distances separating them (Nedelcu, 2019). Based on these comparisons, it has been argued that evolution and complexification of animal forms could have been boosted by the acquisition of the SAND domain at the onset of this lineage through lateral gene transfer; this shared domain of animals, plants and multicellular volvocine green algae would explain why all of these lineages have an exuberant diversity developmental patterns and cell types (Nedelcu, 2019).
Remarks
Taking into account the patchy distribution of these machineries in all the different lineages of early-branching animals, we can infer that the genetic toolkit of the animal LCA was already rich in genes previously thought to be metazoan innovations, from mechanisms to define epithelia to neuron-like signaling cells and muscle-like contractile cells. Despite early-branching lineages have seemingly evolved independent kind of cell types (Steinmetz et al., 2012, Moroz et al., 2015 I think), the cell type-specific domains and domain architectures are present in all their proteomes. The integration of these pathways and mechanisms was thus largely complete in the animal LCA (similar to the observations by Putnam about the cnidarian-bilaterian LCA, 2007), suggesting that those machineries were likely present in an ancestral state, to the least, in the first animals.
Together with other studies that we will discuss in the future, these studies have changed the current consensus: as the majority genes relevant for important developmental processes in all grades of animal complexity have been found in earlier-branching lineages, it is thought that expansion, co-option and (from temporal to spatial) sophistication of the regulation of these genes were the responsible agents for the gradually increasing complexity of animals (Tweedt and Ervin, 2016).
There is one more piece of important information that is missing from the picture. Given that animals are multicellular and a lot of the hustle about the origins of animals is about having many different cells, what can the cells of animals tell us about their origin? We will keep exploring this topic through the lens of single cells in the next post.